An Introduction to Corrosion
Before you can understand why Enviropeel is so successful in preventing corrosion it would be helpful to understand a little about why corrosion occurs, with particular attention to the effects that you will find on projects around the world.
Corrosion is the disintegration of metal through an unintentional chemical or electrochemical action, starting at its surface. The driving force that causes metals to corrode is a natural consequence of their temporary existence in metallic form. The energy that is absorbed and stored during the transformation into a metallic state from the original ore is gradually released through corrosion, returning the metal to its original compounds.
 These pictures illustrate the similarity in color between pale green malachite, a common copper ore mineral, and the corrosion products on a brass plate (70% copper) exposed to a humid environment.
These pictures illustrate the similarity in color between pale green malachite, a common copper ore mineral, and the corrosion products on a brass plate (70% copper) exposed to a humid environment.

Some words crop up all the time in descriptions of corrosion, here are some definitions to help understand them.
Electrolyte: This generally refers to the solution of a chemical compound, such as salt, that dissociates into electrically charged ions when dissolved in a liquid. The resulting electrolyte (or electrolytic) solution is an ionic conductor of electricity.
Atomic structure – the proton, neutron and electron: An atom has a dense central core (the nucleus) consisting of positively charged particles (protons) and uncharged particles (neutrons). Negatively charged particles (electrons) are scattered in a relatively large space around this nucleus and move about it in orbital patterns at extremely high speeds. It is the transfer of electrons during oxidization that lies at the heart of the corrosion process.
Oxidation, anodes and the galvanic series: In a narrow sense, oxidation means the reaction of a substance with oxygen. In the case of iron, the reaction with oxygen causes it to be oxidized to rust, as electrons are transferred from the oxidizing iron to the oxygen. The transfer of the electrons and the formation of the iron oxide is the basic mechanism that produces loss of metal from the original iron. The process is referred to as an anodic reaction.
All metals exhibit a tendency to be oxidized, some much more easily than others. The relative strength of this tendency is recorded in the galvanic series. A metal's position in the series is a key factor in galvanic corrosion effects, where different metals are in contact with one another. Environmental factors, such as oxygen and an electrolyte (usually water) also play a significant role in the speed of the corrosion process
A corrosion cell works like a battery. Exposure of metal to an electrolyte completes a circuit, producing a current flowing from the anode to the cathode.
The surface of a metal, when exposed to an electrolyte, will have some sites that are more vulnerable to an anodic reaction, producing electrons that are consumed at other less vulnerable sites by cathodic reaction. These sites together make up the ‘corrosion cell’.
A Corrosion cell
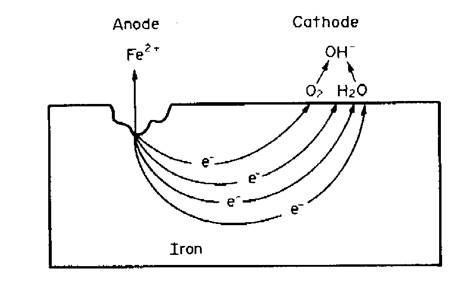
The anodes and the cathodes of the corrosion cell are not necessarily fixed in location - they can be adjacent or widely separated, depending on a number of different factors:
The process occurring at the anodic sites is the dissolution of metal as metallic ions in the electrolyte or the conversion of these ions to insoluble corrosion products such as rust. The flow of electrons between the corroding anodes and the non-corroding cathodes forms the corrosion current, producing the destructive process we see as corrosion.
Uniform Corrosion:Uniform corrosion occurs over the majority of the surface of a metal at a steady and predictable rate. Its predictability facilitates easy control, the most basic method being to make the material thick enough to function for the lifetime of the component. It is responsible for 30% of corrosion failures.
Localised corrosion: Failures from localised corrosion are far less predictable than uniform
corrosion and, because the failure may occur without warning, have consequences that are far more severe. Responsible for 70% of corrosion failures, they can occur after a surprisingly short period of use or exposure. There are a variety of mechanisms for localized corrosion, the most relevant for our training purposes are:
Galvanic corrosion: This can occur when two different metals are placed in contact with each other and is caused by the greater willingness of one to give up electrons than the other. Three special features of
this mechanism need to operate for corrosion to occur:
Pitting corrosion: This occurs when a coating breaks down. A small area of exposed metal gives up electrons easily and the reaction initiates tiny pits with localised chemistry supporting rapid attack. Pits can be crack initiators in stressed components or those with residual stresses resulting from forming operations. This can lead to stress corrosion cracking.
Crevice Corrosion: Crevice corrosion occurs when oxygen cannot penetrate a crevice exposed to an electrolyte and a differential aeration cell is set up. Corrosion occurs rapidly in the area with less oxygen as electrons flow to more oxygenated areas, causing severe material loss.
If we think of corrosion as a natural process which can be affected by a number of factors, we can better understand how, by interrupting this process, corrosion effects can be reduced or eliminated. In the three corrosion mechanisms describe above, just by eliminating the electrolyte, the effects of corrosion would, for all practical purposes, be eliminated. But, as can be seen in the case of pitting corrosion, localized coating failure can then lead to accelerated effects.
In most cases, a good quality coating on a simple surface, such as a steel pipeline, will provide corrosion protection by preventing exposure to oxygen and water-based electrolytic solutions. It is at the joints where the problems occur. Here, with multiple components, widely varying in size, and coating issues to do with assembly and complexity, exposure to corrosion mechanisms is far more likely, and far more difficult to eradicate. Enviropeel’s combination, of a flexible barrier coating providing passive protection and active protection from its built-in corrosion inhibitors, is designed to eliminate all possible causes of corrosion.
Source: http://www.ae-sys.com/techpapers/corrosion%20defined%20for%20training%20doc.doc
Web site to visit: http://www.ae-sys.com/
Author of the text: indicated on the source document of the above text
In this section, a detailed description of the electrochemical processes leading to and controlling corrosion will be outlined. The first section deals with the electrochemical basis for corrosion, which is controlled by thermodynamic principles, while the effect of kinetic, or rate control will be introduced later in the section. It is important to understand the role of both thermodynamic and kinetic processes in initiating and controlling corrosion in order to determine corrosion rates, or reduce corrosion by different methods, such as sacrificial anodes or alloy development such as stainless steels.
In most corrosion processes, with the exception of cathodically controlled processes, metal will dissolve. An understanding of the principles behind how metal dissolves in a solution is a necessary part of understanding corrosion. Consider a piece of pure metal M in a solution which contains its own ions, Mz+. In this case the metal has solubility in the solution in which it is placed. An example would be a piece of pure copper in a solution of copper sulfate.
The general equilibrium reaction for a metal electrode in a solution of its own ions is:-
M = Mz+ + ze-
where M represents a metal atom, Mz+ is the metal ion, Z is the possible valences and e- represents an electron.
At equilibrium there is no net current flow and the above reaction can be separated into two different reactions. One reaction produces metal ions in solution from metal atoms in the solid with the reaction :-
M > Mz+ + ze-
Again at equilibrium there is no net current, so the electrons produced in the reaction above are consumed in a reaction where all the electrons combine with metal ions in solution to deposit as atoms on the solid metal surface in contact with solution in the reaction :-
Mz+ + ze- > M
These individual reactions are called “half cell” reactions as they another reaction to either consume or produce electrons for them to proceed. These half cell reactions are also oxidation and reduction reactions.
Oxidation is a reaction that produces electrons. The ionization of metal atoms to ions is therefore an oxidation half cell reaction.
Reduction is a reaction that consumes electrons. De-ionization to form a metal atom is therefore a reduction half cell reaction.
M = Mz+ + ze-
The above reaction is called a “REDOX” reaction because it contains both an oxidation and a reduction half cell reaction.
Both oxidation and reduction are important reactions. The oxidation reaction is the basis of corrosion as metal is dissolved whilst transforming from atoms to ions in a solution. The reduction reaction is also important as it is the basis of electroplating where ions in solution are deposited as atoms on a surface, for example chrome or gold plating.
During corrosion, the half cell reactions do not usually involve the same elements and ions in both the oxidation and reduction reactions. The electrons produced in the oxidation reaction are not available for the reverse reduction reaction, which would replace the metal atoms back on the metal surface. Instead, the electrons from the oxidation reaction are consumed by a different reduction reaction resulting in a new reaction product. This leads to a secondary reaction between the products of the oxidation and reduction half cells to form a stable material. These can further react with the environment to form stable compounds such as rust. These are then very difficult reactions to reverse back to the atomic form of the metal which is one of the major reasons why corrosion is an extremely deleterious process.
Basic Corrosion Processes.
Four necessary processes are required to have corrosion take place. All these processes must be available for corrosion to occur. If any of these are unavailable then corrosion will not occur. This provides the first measures of corrosion protection – by disabling any one of the four necessary processes, then corrosion will be blocked. This very simple approach to corrosion protection is often overlooked but can be an invaluable tool for the engineer to avoid costly corrosion problems.
Oxidation Half Cell Reaction.
The first process is an oxidation or anodic half cell reaction. The oxidation half cell reaction was described above. The corrosion process is essentially this oxidation reaction as metal atoms are transformed to their ions. Solid metal in a solution is transformed into metal ions in solution, resulting in weight loss or thinning of the solid material.
M > Mz+ + ze-
Reduction Half Cell Reaction.
The second process of a corrosion cell is a reduction or cathodic reaction. One reduction or cathodic reaction was also described above which is the plating or deposition reaction.
Mz+ + ze- > M
Other cathodic reactions are described below in terms of their REDOX reactions, although a cathodic reaction would be in the direction from left to right:-
2. Reduction of hydrogen ions in strong, de-aerated acids:
2H+ + 2e- = H2
For the cathodic half cell reaction hydrogen gas is produced at the same time as the anodic reaction is progressing..
3. Reduction of oxygen in weak aerated acids to form water.
O2 + 4H+ + 4e- = 2H2O
4. Change in ionic state, e.g., Cupric to cuprous ion.
Cu2+ + e- = Cu1+
5. Reduction of oxygen in neutral or basic solutions to form hydroxyl ions.
O2 + 2H2O + 4e- = 4(OH-)
For many practical service situations the last reaction is the most important. All the necessary constituents for the reaction to proceed are present in normal environmental exposure. The oxygen is present as dissolved oxygen in water. A later section will discuss the reduction of oxygen in water in greater detail. The only component missing are four electrons, which during corrosion will be produced by the oxidation or anodic half cell reaction.
Cathode Site.
An important feature at this stage is to distinguish the cathodic half cell reaction from the physical cathode. The physical cathode is the location where the cathode half cell reaction occurs. The cathode only provides the electrical conductivity necessary for electron transfer for the cathode half cell reaction. An example can be obtained from the reduction of oxygen in water reaction above, reaction 5. This reaction will occur on a surface which has the capability to conduct electrons. The surface only needs to be a conductor of electricity, it does not need to be metallic, therefore non metallic materials that are electrically conductive can be cathodes. It should also be apparent that the cathode does not dissolve and can be termed as being “cathodically protected”. Another cathode reaction is reduction of hydrogen ions to hydrogen, reaction 2. In this case hydrogen gas will be nucleated on a conducting surface as a corresponding anodic reaction produces the electrons for consumption. This distinction between cathode reaction and the cathode material is important in later stages when localized corrosion forms such as pitting and crevice corrosion are discussed and large amounts of the metal surface exposed to an environment show no signs of corrosion while extensive corrosion is present in local regions on the surface. It is thought that the non–corroded regions are cathodically protected.
Ion Transport.
The third process is ionic transport for which a conductive medium or electrolyte is required. In many cases of environmental exposure this can be water, seawater, acidic or basic solutions. The greater the ionic flow capability of the solution the greater the corrosion rate which can be supported. Therefore distilled water with a high resistance and low conductivity, usually supports a lower corrosion rate than seawater with a low resistance and high conductivity.
Electron Transport.
The fourth necessary process is a mechanism for electron transport between the anode and cathode sites. As electrons are produced by the oxidation anodic half cell reactions and consumed by the reduction cathodic reaction, electrons flow from the anode to the cathode. Without flow of electrons between the anode and cathode, these half cell reactions cannot occur. Blocking this electron flow is therefore another method of corrosion protection. The anode therefore has an excess of electrons and is therefore the negative pole. The cathode is the positive pole.
The necessary electron flow can be achieved by several mechanisms. One is a wire conductor which connects the anode and cathode. Electrons flow along the wire from the anode to the cathode. A good example of this process is a flashlight. The battery in the flashlight has a cathode and an anode. The zinc casing is the anode and usually the negative pole. The center post of carbon is the positive pole. When the switch is placed in the “on” position a circuit is completer between the positive and negative pole through the light bulb filament which glows white hot due to resistive heating from the electron flow from the anode to the cathode. The electron flow is therefore providing both a voltage and a current.
A second and much more important method of electrical connection for an engineer is controlled by the microstructure of the materials. The intimate contact of phases in a metallic alloy can provide the necessary electrical contact to establish anode and cathode regions. It should be remembered that each phase has its own distinct composition so each phase will react differently to the environment. These differences will cause anodes and cathodes to form which are in physical contact. Impurities in metals can also act as anodes or cathodes due to chemical differences.
In summary, the four necessary processes for a corrosion cell are anode reaction, cathode reaction, electrolyte and electron transport. Unfortunately for many engineering materials such as steels in water, all these four processes are present and corrosion will occur. For other materials in the same environment, no corrosion will occur. An example would be gold in water. If there is no corrosion, then one of these four processes is not occurring, For gold it so happens that no anode half cell reaction is available which makes it immune to corrosion.
Exchange Current Density.
The REDOX half cell reactions of oxidation and reduction both involve electrons. The oxidation, anodic reaction produces electrons while the reduction, cathodic reaction consumes them.
M = Mz+ + ze-
At equilibrium for a REDOX reaction there is no net ionic or electron flow, so there is no corrosion weight lost by the metal or deposition or weight increase of the metal. The weight of the metal M will be the same at the end as at the beginning and the concentration of Mz+ in solution will be the same at the end as the starting concentration. However, for an oxidation reaction, there is a current flow –number of electrons per unit time- as they are produced in the anodic half cell reaction. There is also current flow for the reduction half cell reaction as electrons are consumed.
M > Mz+ + ze-
Mz+ + ze- > M
At equilibrium the number of electrons produced by the anodic half cell reaction and the number of meal ions transformed from metal atoms is the same as the number of electrons consumed by the cathodic half cell reaction and also the number of ions transformed back to atoms, and so both the half cell reactions proceed at equal rates. The current flows for the anode and cathode half cell reactions are finite and identical, but in opposite directions. But the magnitude of the current flows is identical for both half cell reactions. This magnitude of current is called the Exchange Current Density. The units used are current per unit area of surface exposed, for example A/cm2, The reaction surface and ions involved controls the exchange current density. Some examples are given below for zinc when it is placed in a solution containing zinc ions of the type indicated in the table below:-
Ion ECD (A/cm2)
Perchlorate 3 x 10-8
Sulfate 3 x 10-5
Chloride 3 x 10-4
The exchange current density or ECD represents an equilibrium point. The metal in solution of its own ion type can only corrode or have ions deposit when the current density is not at the exchange current density value.
Potentials.
As described above, REDOX reactions are both anodic and cathodic half cell reactions but balanced under equilibrium conditions. Clearly, some standard has to be set for this equilibrium and so standard conditions are defined as one atmosphere of gas pressure, pure solid present, normal ionic solution strengths at 25oC temperature. Normalcy is the number of equivalents per liter of solution. Normal ionic strength is when one gram equivalent is dissolved per liter of water. For hydrochloric acid, a one normal solution would be the gram molecular weight, or gms per liter of solution. For one normal sulfuric acid, H2SO4, half the gram molecular weight in a liter of water would be required as it has two hydrogen ions present and a normal solution requires only one electron to be available. For basic solutions the number of hydroxyl ions is used instead of hydrogen ions or the number of electrons transferred in the reaction.
What is unavailable at this stage is a scale to differentiate between the different REDOX reactions listed for both metals and non metals. A reference scale is required. This is sometimes called the Electro Motive Force (EMF) series or REDOX potential series.
To measure potentials (or voltages) for comparison of REDOX reactions, the reference used is a Standard Hydrogen Electrode (SHE). This electrode is a constructed by bubbling gaseous hydrogen at 1 atm. of pressure over a piece of pure platinum in a solution of one normal H+ ions. The redox reaction is therefore:-
2H+ + 2e- = H2g

Standard Hydrogen Electrode.
The potential for this reaction by definition is 0.0 V. It is a complete REDOX reaction and will maintain a standard, constant voltage unless the conditions change away from the defined standards. The oxidation, anodic reaction and the reduction, cathodic half cell reactions are given below:-
H2 > 2H+ + 2e-
2H+ + 2e- > H2
No electron removal is allowed and the hydrogen ion strength must be maintained.
By coupling different metals M to the SHE their potentials can be measured in volts on a voltmeter. This gives rise to the data below for EMF or REDOX potentials. Note that is some areas , the scale is opposite with gold for example at –1.48V. Most engineering practices use the scale below, but be careful.
Reaction Potential at equilibrium (volts).
Au = Au3+ + 3e- 1.498
O2 + 4H+ + 4e- = 2H2O 1.229
Pt = Pt2+ + 2e- 1.2
O2 + 2H2O + 4e- = 4(OH-) +0.401
2H+ + 2e- = H2 0.0
Fe = Fe2+ + 2e- -0.440
Cr = Cr3+ + 3e- -0.744
Zn = Zn2+ + 2e- -0.763
Al = Al3+ + 3e- -1.662
In the figure below showing how a SHE is coupled to the REDOX reaction being measured, a high resistance voltmeter is used to measure the voltage for this series to ensure that no current flows from the REOX cells and so no corrosion or deposition is occurring as all the reactions are equal and opposite in rate. The REDOX reaction being measured must also be at standard condition. This series provides the thermodynamic probability that a pure metal under standard conditions will either ionize by an anodic reaction or plate out in a cathodic reaction.
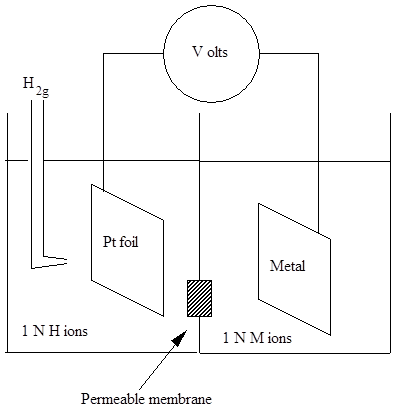
Cell for determination of EMF series or Redox potentials.
The more negative the potential measured in the EMF or REDOX series, the more likely the material is to oxidize in an anodic half cell reaction. Therefore, gold, Au is termed very noble as it has a high positive EMF value. Zinc is very anodic as it has a low negative value of EMF. Therefore connecting a half cell of gold to a half cell of zinc will induce anodic reaction of the zinc to produce electrons, i.e.:-
Zn -> Zn2+ + 2e-
Concurrently, the cathodic half cell reaction will be favored in the half cell with the more positive potential and gold ions will be reduced to gold by:-
Au3+ + 3e- -> Au.
The zinc will have corroded as weight loss will occur. This process is essentially galvanic corrosion. As will be discussed later this is a useful corrosion process as protection techniques sometimes rely on dissolving a cheap, easily replaced metal rather than the expensive structural metals. Two examples of this are galvanizing and sacrificial anodes.
Some important features for the SHE and EMF series.
The standard hydrogen electrode is a difficult piece of apparatus to use. Hydrogen gas is required along with a reasonably strong acid solution. It is not practical for everyday use. Secondly the EMF series only applies to pure metals in highly specified conditions, which are not often seen, such as one normal ionic solutions. It is a good guide only for these conditions. Thirdly, some elements are very low in the series such as chromium, yet this element is added to iron to make steels "stainless".
At this point there is some discrepancy between theory and practice, which is answered by the so far un-described role of kinetics on corrosion. Kinetics involves the rate of a reaction; thermodynamics considers whether a reaction is favorable or not. For a reaction to proceed it must be thermodynamically favorable. So reactions can be thermodynamically favorable, but kinetics determines the rate at which the reaction proceeds. Fortunately in corrosion, the kinetics of a reaction will often aid in reducing the expected rate. Examples would be stainless steels and titanium and its alloys.
Reference Electrodes.
The Standard Hydrogen reference electrode has already been described in earlier notes. It is an impractical electrode with a strong acid and highly flammable hydrogen gas required. A simpler reference electrode is required for practical measurements and several exist. The most common ones are the Calomel electrode, the copper/copper sulfate electrode and the silver/silver chloride electrode.
The requirements of a reference electrode is that are a constant reference against the standard hydrogen electrode over a wide range of conditions. This usually means that a self contained half cell reaction occurs that cannot be change by the environment in which the reference electrode is placed. A good example is the Saturated Calomel Electrode or SCE. A diagram of the electrode is shown below.

Saturated Calomel Reference Electrode.
This is a very common electrode. It is based on the mercurous to mercury half cell reaction of:-
Hg2Cl2 = Hg2++ + 2Cl-
Hg2++ + 2e- = 2Hg
Overall Reaction:-
Hg2Cl2 + 2e- = 2Hg + 2Cl-
Nernst Equation gives:-
ECal = Eo - (RT/zF) ln (aCl-)2
= Eo - (2.303RT/F) log aCl-
ECal = +0.2677 - 0.059 log aCl-
Ecal = +0.242 V against SHE for
Hg/Hg2Cl2 in saturated KCL.
In the SCE the Calomel paste of mercurous chloride is mixed with mercury and and the filling solution is saturated potassium chloride. The saturated solution has a fixed chloride ion activity and the mercurous chloride is only slightly insoluble in water. A porous tip enables ionic transport to complete the necessary components for measuring voltage along with connection to a high resistance voltmeter.The electrode maintains its chemical conditions and provides a reference potential against the standard hydrogen electrode of +0.242 V.
Other common reference electrodes are the Copper/Cupric Sulfate electrode where a copper bar is placed in a saturated cupric sulfate solution.
E Cu/CuSO4 = +0.34 V(SHE) for saturated copper sulfate solution
So far, much has been examined regarding equilibrium conditions. However, corrosion and deposition do not occur under equilibrium conditions. The measurement of the corrosion rate of material in the environment is vital to the correct selection of a material. One method to place the material in the environment and monitor any weight change. Another is conduct a laboratory study and conduct measurements. The issue is what should be measured. In this section, the parameters which provide information on corrosion rates and which can be measured in a laboratory will be determined.
In considering the general REDOX reaction at a metal electrode - M :-
M = Mz+ + ze-
At the Redox potential under standard conditions used to determine the Redox series, there is no net current flow as the anode reaction rate is the same as the cathode reaction rate. This voltage will be called Er for Redox potential. When the potential moves away from this equilibrium value so it no longer equals Er, then the reaction rates becomes unbalanced and one or other of the half cell reaction dominates, either cathodic or anodic. At this new potential either a net flux of electrons will be produced, if the anodic half cell dominates, or electrons will need to be provided to allow the cathodic half cell reaction to proceed. So by controlling the potential the reaction can be either anodic or cathodic. This is also called Polarization, the movement away from the Redox potential value.
Polarization of an Electrode.
Polarization of an electrode is the movement away from the equilibrium state where the anode reaction rate is equal and opposite to the cathodic rate at standard conditions. A simplified model of the situation will be presented in this section to determine what happens when the conditions are altered away from the standard ones. The particular case will be when some electrons produced by the anodic half cell reaction are removed and not available for use by the cathodic half cell reaction.
Consider a pure metal at equilibrium in its own ions. Atoms sit in low energy positions on the surface. An activation energy barrier exists for these atoms to become ions and produce electrons. A schematic diagram of the situation is shown in the first figure below.
At equilibrium the ion exchange current density is available of atoms transforming to ions (anode reaction) and at an equal rate the ions from solution are transforming to atoms (cathodic reaction) at the redox potential of Er
At equilibrium the activation energy barrier is DGe
DGe = BF
The energy barier, DGe, is the same independent of the reaction direction, anodic or cathodic.
In the second figure below, the energy of the atom in the surface was increased, or it was Anodically Polarised to make it more favorable for the atoms to form ions.
Assume BE = FG
Let BC/AC = a
Then AB/AC =1-a
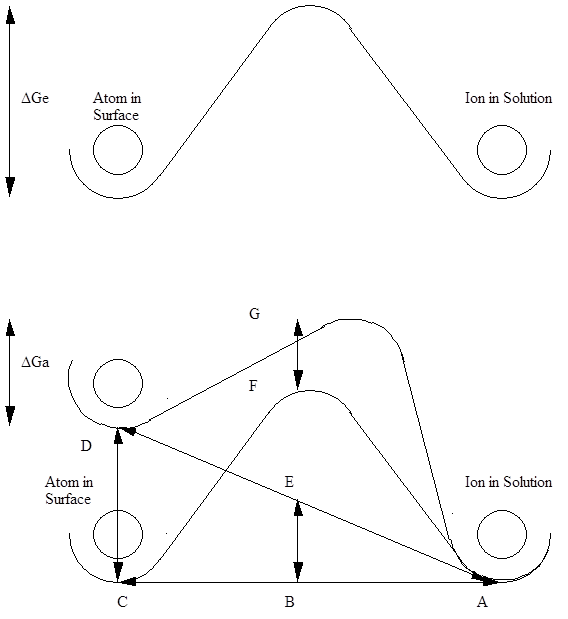
By similar triangles, CD/EB = AC/AB =1/(1-a)
EB = CD(1-a)
CD = Change in anodic energy applied by an external voltage
CD = zhaF
where z - number of electrons involved in the half cell reaction,
ha - overpotential or voltage movement from Er value, F - Faradays constant (96500 coulombs).
EB = (1-a)zhaF
Then the new activation energy barrier for an atom to ion transfer, releasing electrons is:-
DGa = DGe - CD + FG
DGa = DGe -CD + EB
DGa = DGe - zhaF + (1- a)zhaF
DGa = DGe - azhaF
Also, as the process is activation controlled it follows an Arhenius rate law of:-
Rate = Ke(-DG/RT)
R - Boltzmanns constant of 1.96 cal/oK, T temperature in oK
The rate in this case is proportional to the current flowing.
Then:-
i = Ke(-DG/RT)
ia= Ke(-DGa/RT)
ia=Ke(-(DGe-azhaF)/RT)
At equilibrium io is the Exchange Current Density at Redox potential, Er
io=Ke(-DGe/RT)
Expanding the equation for anodic dissolution:-
ia= K exp(-DGe/RT)exp(azhaF/RT)
ia = ioexp(azhaF/RT)
Taking logs
ln ia = ln io + azhaF/RT
Rearranging:-
ha = (ln ia - ln io)(RT/azF)
For cathodic polarization.
hc = (ln io - ln ic)((RT/(1-a)zF)
These are the Tafel equations for polarization of an electrode. They follow the general form of a straight line if h is plotted as a function of log i.
y= c + mx
h = a +/- b logi
The equation above is the TAFEL equation which relates potential to current density for an electrochemical reaction.
The constants for the equation are listed below. The constant "b" is called the Tafel constant.
Anodic
a = -ln io(RT/azF) b = RT/azF
Cathodic
a = ln io(RT/(1-a)zF) b= -(RT/(1-a)zF)
Overall ,changing the conditions from the half cell reactions therefore produces polarization resulting in a current.
Source: http://che.uri.edu/course/CHE534w/Chap%202.doc
Web site to visit: http://che.uri.edu/
Author of the text: indicated on the source document of the above text
GENERAL CORROSION DUE TO ENVIRONMENTAL EXPOSURE.
Uniform Corrosion.
During uniform corrosion the complete exposed surface of the metal corrodes. A good example would be the formation of rust on unprotected steel such as unpainted automobile bodies. This type of corrosion is very general and resulted in a great loss of metal and money.
Uniform corrosion was occurring on this tank when the paint system did not provide complete protection. A considerable amount of rust was found on the surface where the paint had broken down and corrosion occurred.
Uniform corrosion is basically thermodynamically controlled, with the Redox potentials and the Nernst equation dictating the process until concentration polarization takes place when the transport of oxygen is limited. As noted previously for the case of iron, the anodic reaction is:-
Fe -> Fe2+ + 2e-
while the cathodic reaction is oxygen reduction to form hydroxyl ions:-
O2 + 2H2O + 4e- -> 4(OH-)
As the whole surface of steel corrodes the anodic sites and cathodic sites are the same surface.
A further example is shown for a steam return pipe. In this case, water condensed in the pipe and uniform corrosion occurred on the inside of the pipe. As the pipe thinned from the inside, enough material was removed until the internal diameter of the threads on the bottom of the pipe was reached. Eventually a drip was found at the joint when the threads were corroded away and a tight seal was no longer possible. This is a long term process.
Another case of uniform corrosion is zinc dissolving in hydrochloric acid. The zinc dissolves with the formation of hydrogen gas. The oxidation of zinc to zinc ions is the anodic reaction and the cathodic reaction is the reduction of hydrogen ions to hydrogen gas.
Zn + 2HCl = ZnCl2 + H2
Note that this reaction is both chemically and electrochemically balanced.
Anode:- Zn -> Zn2+ + 2e-
Cathode:- 2H+ +2e- -> H2
The rate of reaction is controlled by the slowest anodic or cathodic process. For example, concentration polarization controls the cathodic reduction of oxygen to hydroxyl ions. This rate is a function of velocity and so faster flowing solutions will corrode iron at a faster rate. Temperature is also a factor in activation controlled corrosion. Raising the temperature will also increase the corrosion rate as the activation energy decreases with temperature. Environment is important. Chloride containing solutions will increase the rate of corrosion over non chloride containing solutions. Instead of Fe2+ reacting with only hydroxyl ions it can now react with chloride ions. Most common metals are very soluble in chloride form and so the reaction is increased.
Materials.
Most metals which do not passivate and which have potentials lower than the Redox potential for reduction of oxygen in water will corrode in water. Examples are steels, zinc, and magnesium. Passivators such as stainless steels and nickel containing alloys such as monels and superalloys do not corrode in water.
Environment.
Data on corrosion rates for a variety of metals is readily available. In general a large range of corrosion rates is obtained depending on the specific exposure conditions. For example, corrosion rates from 0.03 mpy in a polar environment to 17.4 mpy at a location at ground level, 60 yards from the ocean at Cape Kennedy for carbon steel were measured. On the beach at Cape Kennedy, the corrosion rate was 42 mpy. In flowing seawater, rates of 15 mpy in the splash zone and 5 to 10 mils just below the surface were reported. From a design aspect, the local conditions of the material must be known to determine thickness to be used for the lifetime of the component. For metals and alloys such as nickel aluminum bronzes, rates of 1 to 2 mpy in quiet seawater were reported.
In designing a piling in a river from unprotected steel, if both sides are exposed to slow flowing water, a 15 mpy rate can be used. For both sides exposed, this becomes 30 mpy of steel lost. For a configuration such as an “I” beam, for a twenty year life expectancy, 600 mils or thousands of an inch of steel will be lost. This does not include the amount of material required for the loading from the decks on the piling which must still be present after twenty years to provide structural stability.
Protection techniques.
1 Exclude the environment.
One of the required components is an electrolyte, which for water also contains the cathodic reactant of oxygen. If this component can be excluded from the surface, then corrosion will not occur. The simplest method is to paint the material. However, this is not always a long lasting solution if the paint is not applied well. The introduction shows a possible problem where the paint will not provide a long term solution.
2. Sacrificial Anodes.
Apply a more active metal. A good example would be zinc on steels. It must be noted that there is a concept called "throwing power" in that the kinetics of the reactions are such that a small piece of zinc cannot protect a large area. Zincs have to be placed at frequent distances with a higher density at increased velocity areas. Cadmium plating on steel was a good choice but it along with chromates are deemed to be environmentally “unfriendly”. These materials are being replaced.
3. Impressed Current techniques
These make the component to be protected a cathode by supplying electrons from an external source.
4. Material selection.
Weathering steels are a steel with 0.5 - 1.0% Cu additions. The copper stabilizes the oxide product on the surface such that after the initial oxidation, the rust formed becomes protective and no further metal penetration occurs. Examples include many light poles throughout the USA. These are form a corrosion perspective, maintenance free structures. The drawback is the only color available is rust!. Other materials which will not corrode can be selected.
Weathering steel light pole, no maintenance of the pole is required.
5. Inhibitors.
Examples of inhibitors are the waxy brown paper surrounding tools. In the paper is a vapor phase inhibitor which coats the metal and displaces the moisture necessary for corrosion.
Testing Technology.
Salt Spray Testing.
Salt spray testing in a chamber is a recognized test for uniform corrosion. It follows the ASTM B117 standard. A 5% by weight salt solution at 100% humidiy and 95F is passed into a chamber in which the samples are contained. The equipment is shown below.
An example of results of a test is shown for two steels, one of which is suitable and one which exhibited extensive rust over the same time period.
On the left of the figure is a typical untested component, the center one corroded while the one on the right was covered on a white deposit from the salt. There are many variations on the salt spray test with each particular industry setting up exposure limits based on their experience. Some groups use 1000 hours with no corrosion, others 24 hours with no “white rust” for galvanized parts. For painted parts an “X” scratch is often placed through the paint into the substrate and in addition to damage in the scratch, damage spreading away from the scratch monitored. This is known as “undercutting”. Other cycles and combinations are used, such as some time in salt spray, followed by a period at elevated temperature and high ultra violet light exposure, followed by a time below freezing. These conditions were defined on the basis that they reflect accurately an acceleration of the failure mechanisms that lead to failure of the particular component.
Another process to measure uniform corrosion is the Tafel extrapolation shown previously. From this test corrosion rates can be established to put into design calculations for the corrosion life of a component. For example, if a corrosion rate of 15mpy is established, or 15 thousands of an inch of metal thickness will be removed each year, the thickness of metal required for a 30 year corrosion life as a piling in a river would be 0.6 inch as water is on both sides of the component.
A case of uniform corrosion is the corrosion of reinforcing steel in concrete. Steel is placed in concrete to reinforce it as concrete is a brittle material in tension, so the steel reinforces it. If the Pourbaix diagram for iron is examined, Chapter 2, then the conditions of concrete a high pH, are such that a passive film should be present for steel in concrete. However, actions such as salting the roads to prevent ice forming, or the local areas proximity to a marine environment, results in salt building up in the concrete cover overlaying the steel reinforcing bar. It is this concrete cover that protects the steel from environmental exposure and uniform corrosion. The chloride ion from salt gradually permeates down to the level of the steel and breaks down the passivation. Once the passivation is destroyed then uniform corrosion of the steel then occurs. The corrosion products require a volume of space considerably greater than the original steel, from 4 to 10 times the volume. This produces tensile stresses in the concrete cover separating the rebar form the external environment. Once the stresses reach the strength of the concrete, it “spalls” and the concrete separates from the structure leaving the reinforcing steel exposed, which then continues to corrode. A good example of the process is shown below.
The possible solutions to this include cathodic protection of the steel, adding inhibitors to the concrete, change the composition of the reinforcing steel, coatings on the reinforcing steel, and non metallic reinforcement materials. This problem is worldwide and
very costly.
In the figure below, a steel wheel nut on a boat trailer has corroded uniformly.
On the left hand side of the nut a large piece of rust is still attached. The rust contains many cracks and is about to fall from the surface. The exansion of corrosion products can be seen here. The steel bearing is on the left hand side and rust containing cracks can be seen as well. The area of concern with an article like a boat trailer is that this rust will build up in the leaf springs rather like a wedge of material and separate them over time.
Galvanic Corrosion.
Galvanic corrosion results from two different metals being in contact in the environment. Examples would be brass plumbing fittings on a cast iron pipe. In this case several reactions are possible, but in general the corrosion rate of the most anodic or active metal is increased and the corrosion rate of the more cathodic metal is decreased. This is shown in the following diagrams. The first figure shows the corrosion rate for a single metal corroding in an environment. A good example would be for zinc in hydrochloric acid. The anode reaction favored is:-
Zn -> Zn2+ + 2e-
2H+ +2e- -> H2
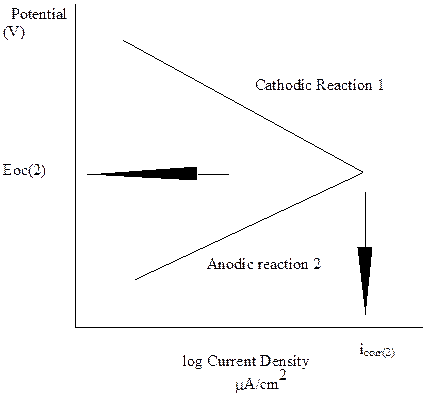
Corrosion rate determination for a two Redox reaction system
Using Faradays Law, substitution of the value of icorr will calculate the corrosion rate in mils per year. The next figures shows how to calculate the oveall corrosion rate as well as the individual reactions rates for each half cell reaction when a third Redox reaction is added at a potential between the first two Redox reactions. The rule that must be applied is that the TOTAL OXIDATION RATE MUST EQUAL THE TOTAL REDUCTION RATE. In figures below, the dashed lines represent the TOTAL rates.
Er(3)
Eoc(2)
icorr(2)

Corrosion rate determination for a three Redox reaction system. The corrosion rate for electrode 2 has increased from icorr to icorr 1+2 as it the only anodic reaction.
Two cases are shown, the first in the figure above, when the corrosion potential for three electrodes, Eoc(3), is above the two electrode potential Eoc(2), and in the figure below, when the three electrode corrosion potential is below the two electrode potential.
In the figure above, the open circuit potential for the three Redox system, Eoc (3) is more negative than the individual third electrode Reox potential, Er(3). As such the third Redox reaction can only contribute to the cathodic reaction rate. The third Redox reaction is therefore protected from corrosion. The total reduction rate is provided by the addition of the cathodic rates from Redox reactions 1 and 3, which is shown by the dashed line. The second Redox dissolution rate increased significantly from icorrr(2) to icorr(3) by the introduction of the third Redox cathodic reaction increasing the total cathode reaction which must be matched by the anode reaction from Redox reaction 1.
In the figure below, a similar process can be followed for determination of corrosion rates. In this case the open circuit potential from the three Redox reactions, Eoc(3) is more positive than the potential for two Redox reactions. In this case both the second and third anodic Redox half cell reactions are proceeding and both will be corroding, but the third electrode is at a lower rate than if the second electrode was not present.

The introduction of a less noble metal will decrease the corrosion rate of the more noble metal.
Both these figures show that introducing a more anodic metal will decrease the corrosion rate for a more noble metal. This is the process behind galvanic corrosion. It can also be used for protection by galvanizing.
Factors Affecting Galvanic Corrosion.
1. Area Effect.
When current flows between the anode and cathode, the CURRENT will be the same in the anode and cathode independent of the surface area of each electrode. It is the CURRENT rather than the CURRENT DENSITY which is equal for the anodic and cathodic reactions. Therefore, if the current flowing between the anode and the cathode is one amp and the surface areas are one cm2, then the current density in each electrode is one A/cm2. However, if the area of the anode is only 0.1 cm2, then the current density in the anode with the same one amp flowing is 10 A/cm2. From Faradays Law, the corrosion rate depends on the CURRENT DENSITY in the anode. In this case decreasing the surface area of the anode increases the corrosion rate by a factor of 10.
As a general rule to minimize galvanic corrosion, the anode area should be large and the cathode area should be small. This leads to a result that on first appearance would seem incorrect. For protection from galvanic corrosion, the CATHODE of the system should be painted if a coating is applied. This arises from the area effect, in that if the paint is damaged by a scratch for example, then a small cathode to large anode area ratio is formed which results in minimizing corrosion rates. If the anode is painted, then damage to the paint results in a large cathode to small anode ratio which results in large corrosion rates in the anode and rapid penetration into the metal.
The anode to cathode area effect is an important characteristic. It is important in several other forms of corrosion including pitting corrosion, crevice corrosion, stress corrosion cracking and corrosion fatigue. Remember the important effects of the anode to cathode area ratios.
2. Physical Distance Effect.
Another important factor for galvanic corrosion is the distance effect. Galvanic corrosion rates are the largest at the interface between the anode and cathode and decrease with distance away from the contact region. As the anodes and cathodes are both good conductors - remember carbon can cause galvanic corrosion by acting as a cathode - electron transport is very good. What is more difficult is the ionic transport in the electrolyte. As the distance between the anodic reaction site and the cathode reaction site increases the transport of the ions becomes more difficult and the corrosion rate decreases. Essentially the resistance between the anode and cathode increases with distance.
This is an important factor in determination of the form of corrosion. If a suspicion of galvanic corrosion exists then it should follow the rule of the corrosion rate being higher adjacent to the galvanic contact region. If the corrosion rate is greater away from this area then another type of corrosion may be involved. For example if corrosion appears at a constriction some distance from a galvanic contact, then erosion corrosion was the cause not galvanic contact.
3. Distance Apart in the Galvanic Series.
A listing of the corrosion resistance in seawater for many metallic systems was produced and it is called the galvanic series. It is an empirical listing of the corrosion resistance of metals. Its advantage over the Redox series is that it refers to alloys in a real environment. Metal at the top of the system are highly cathodic while metals at the bottom highly anodic.
For selection purposes metals close together on the list are desirable as there is little driving force for corrosion to be accelerated.
Clearly, it is undesirable to connect metals widely spaced on the galvanic series.
4.Temperature Effects.
With increasing temperature above 180oF, zinc will form a protective layer and become cathodic to iron. The zinc becomes nonprotective and aggressive to the iron.
It is best to conduct a test program to determine the relative corrosion rates for the metals under the conditions they will be exposed to in order to determine any unusual effects which may take place due to unpredicted reactions.
Prevention of Galvanic Corrosion.
1. Electrically Insulate the Metals.
Electrical insulation of the dissimilar metals will avoid galvanic corrosion. One method demonstrated in the class is to use non metallic spacers.
2. Materials Selection.
Try to avoid selecting dissimilar metals. If that cannot be avoided refer to some of the following protection techniques.
3. Design.
Several features can be designed in to minimize the effects of
galvanic corrosion:-
a) Design for the cheapest replacement.
Allow corrosion to occur in a component that can be replaced without loss of production capability. This may mean some redundant systems such as multiple valves and pipes in one area. However, this would allow one pipe system to be used as a bypass while the corroded pipe from the first system is replaced without loss of production. After the second pipe exhibits corrosion then use the first pipe system and replace the second pipe. This is corrosion control rather than prevention. However, there is no plant downtime and the corroded component is a piece of pipe rather than an expensive and difficult to replace valve.
b) In sealed systems use the area effect.
In a sealed system where the supply of dissolved oxygen for the cathodic reaction is limited and easily expended, if a large anode to small cathode area is designed then only a very small degree of corrosion over the large anode will occur before the oxygen consumed. Then no more corrosion will occur. If a brass fitting is placed on a water tank fro a closed system where the steel surface area of the tank is much larger the brass area, then the steel will corrode slightly until all the oxygen is used up and then it will cease.
4 Coatings.
Paint the cathode to prevent the cathodic reaction.
5. Sacrificial Protection
The addition of a sacrificial element will avoid corrosion .
6. Impressed Current Protection.
If electrons are supplied to the anode then it will not corrode despite the galvanic contact with a more nobler metal.
7. Inhibitors.
Cars use inhibitors in antifreeze as there is often galvanic contact in the water cooling system between the aluminum alloy cylinder head and the cast iron engine block and water pump.
Testing Technology.
Source: http://che.uri.edu/course/CHE534w/Chap%204.doc
Web site to visit: http://che.uri.edu
Author of the text: indicated on the source document of the above text
1. General
The US Federal Highway Administration released a study, entitled Corrosion Costs and Preventive Strategies in the United States, in 2002 on the direct costs associated with metallic corrosion in nearly every U.S. industry sector. The study showed that for 1998 the total annual estimated direct cost of corrosion in the U.S. was approximately $276 billion (approximately 3.1% of the US gross domestic product). FHWA Report Number:FHWA-RD-01-156. The NACE International website has a summary slideshow of the report findings. Jones1 writes that electrochemical corrosion causes between $8 billion and $128 billion in economic damage per year in the United States alone, degrading structures, machines, and containers.
Corrosion involves the deterioration of a material as it reacts with its environment. Corrosion is the primary means by which metals deteriorate. Corrosion literally consumes the material reducing load carrying capability and causing stress concentrations. Corrosion is often a major part of maintenance cost and corrosion prevention is vital in many designs. Corrosion is not expressed in terms of a design property value like other properties but rather in more qualitative terms such as a material is immune, resistant, susceptible or very susceptible to corrosion.
The corrosion process is usually electrochemical in nature, having the essential features of a battery. Corrosion is a natural process that commonly occurs because unstable materials, such as refined metals want to return to a more stable compound. For example, some metals, such as gold and silver, can be found in the earth in their natural, metallic state and they have little tendency to corrode. Iron is a moderately active metal and corrodes readily in the presence of water. The natural state of iron is iron oxide and the most common iron ore is Hematite with a chemical composition of Fe203. Rust, the most common corrosion product of iron, also has a chemical composition of Fe2O3.
The difficulty in terms of energy required to extract metals from their ores is directly related to the ensuing tendency to corrode and release this energy. The electromotive force series (See table) is a ranking of metals with respect to their inherent reactivity. The most noble metal is at the top and has the highest positive electrochemical potential. The most active metal is at the bottom and has the most negative electrochemical potential.
Corrosion involve two chemical processes…oxidation and reduction. Oxidation is the process of stripping electrons from an atom and reduction occurs when an electron is added to an atom. The oxidation process takes place at an area known as the anode. At the anode, positively charged atoms leave the solid surface and enter into an electrolyte as ions. The ions leave their corresponding negative charge in the form of electrons in the metal which travel to the location of the cathode through a conductive path. At the cathode, the corresponding reduction reaction takes place and consumes the free electrons. The electrical balance of the circuit is restored at the cathode when the electrons react with neutralizing positive ions, such as hydrogen ions, in the electrolyte. From this description, it can be seen that there are four essential components that are needed for a corrosion reaction to proceed. These components are an anode, a cathode, an electrolyte with oxidizing species, and some direct electrical connection between the anode and cathode. Although atmospheric air is the most common environmental electrolyte, natural waters, such as seawater rain, as well as man-made solutions, are the environments most frequently associated with corrosion problems.
A typical situation might involve a piece of metal that has anodic and cathodic regions on the same surface. If the surface becomes wet, corrosion may take place through ionic exchange in the surface water layer between the anode and cathode. Electron exchange will take place through the bulk metal. Corrosion will proceed at the anodic site according to a reaction such as
M → M++ + 2e-
where M is a metal atom. The resulting metal cations (M++) are available at the metal surface to become corrosion products such as oxides, hydroxides, etc. The liberated electrons travel through the bulk metal (or another low resistance electrical connection) to the cathode, where they are consumed by cathodic reactions such as
2H+ + 2e- → H 2
The basic principles of corrosion that were just covered, generally apply to all corrosion situation except certain types of high temperature corrosion. However, the process of corrosion can be very straightforward but is often very complex due to variety of variable that can contribute to the process. A few of these variable are the composition of the material acting in the corrosion cell, the heat treatment and stress state of the materials, the composition of the electrolyte, the distance between the anode and the cathode, temperature, protective oxides and coating, etc.
Types of Corrosion
Corrosion is commonly classified based on the appearance of the corroded material. The classifications used vary slightly from reference to reference but there is generally considered to be eight different forms of corrosion. There forms are:
Uniform or general – corrosion that is distributed more or less uniformly over a surface.
Localized – corrosion that is confined to small area. Localized corrosion often occurs due to a concentrated cell. A concentrated cell is an electrolytic cell in which the electromotive force is caused by a concentration of some components in the electrolyte. This difference leads to the formation of distinct anode and cathode regions.
Intergranular – preferential corrosion at or along the grain boundaries of a metal.
Galvanic – corrosion associated primarily with the electrical coupling of materials with significantly different electrochemical potentials.
Environmental Cracking – brittle fracture of a normally ductile material that occurs partially due to the corrosive effect of an environment.
Erosion corrosion – a corrosion reaction accelerated by the relative movement of a corrosive fluid and a metal surface.
Fretting corrosion – damage at the interface of two contacting surfaces under load but capable of some relative motion. The damage is accelerated by movement at the interface that mechanically abraded the surface and exposes fresh material to corrosive attack.
Dealloying – the selective corrosion of one or more components of a solid solution alloy.
Dezincification – corrosion resulting in the selective removal of zinc from copper-zinc alloys. Erosion corrosion – a corrosion reaction accelerated by the relative movement of a corrosive fluid and a metal surface.
Fretting corrosion – damage at the interface of two contacting surfaces under load but capable of some relative motion. The damage is accelerated by movement at the interface that mechanically abraded the surface and exposes fresh material to corrosive attack.
Dealloying – the selective corrosion of one or more components of a solid solution alloy.
Corrosion is deterioration of essential properties in a material due to reactions with its surroundings. In the most common use of the word, this means a loss of an electron of metals reacting with water or oxygen. Weakening of iron due to oxidation of the iron atoms is a well-known example of electrochemistry (a branch of chemistry that studies the reactions that take place when an ionic and electronic conductor interfere) corrosion. This is commonly known as rust. This type of damage usually affects metallic materials, and typically produces oxide(s) and/or salt(s) of the original metal. Corrosion also includes the dissolution of ceramic materials and can refer to discoloration and weakening of polymers by the sun's ultraviolet light.
Most structural alloys corrode merely from exposure to moisture in the air, but the process can be strongly affected by exposure to certain substances (see below). Corrosion can be concentrated locally to form a pit or crack, or it can extend across a wide area to produce general deterioration. While some efforts to reduce corrosion merely redirect the damage into less visible, less predictable forms, controlled corrosion treatments such as passivation and chromate-conversion will increase a material's corrosion resistance.
Most ceramic materials are almost entirely immune to corrosion. The strong ionic and/or covalent bonds that hold them together leave very little free chemical energy in the structure; they can be thought of as already corroded. When corrosion does occur, it is almost always a simple dissolution of the material or chemical reaction, rather than an electrochemical process. A common example of corrosion protection in ceramics is the lime added to soda-lime glass to reduce its solubility in water; though it is not nearly as soluble as pure sodium silicate, normal glass does form sub-microscopic flaws when exposed to moisture. Due to its brittleness, such flaws cause a dramatic reduction in the strength of a glass object during its first few hours at room temperature.
The degradation of polymeric materials is due to a wide array of complex and often poorly-understood physiochemical processes. These are strikingly different from the other processes discussed here, and so the term "corrosion" is only applied to them in a loose sense of the word. Because of their large molecular weight, very little entropy can be gained by mixing a given mass of polymer with another substance, making them generally quite difficult to dissolve. While dissolution is a problem in some polymer applications, it is relatively simple to design against. A more common and related problem is swelling, where small molecules infiltrate the structure, reducing strength and stiffness and causing a volume change. Conversely, many polymers (notably flexible vinyl) are intentionally swelled with plasticizers, which can be leached out of the structure, causing brittleness or other undesirable changes. The most common form of degradation, however, is a decrease in polymer chain length. Mechanisms which break polymer chains are familiar to biologists because of their effect on DNA: ionizing radiation (most commonly ultraviolet light), free radicals, and oxidizers such as oxygen, ozone, and chlorine. Additives can slow these process very effectively, and can be as simple as a UV-absorbing pigment (i.e., titanium dioxide or carbon black). Plastic shopping bags often do not include these additives so that they break down more easily as litter.
The remainder of this article is about electrochemical corrosion.
One way to understand the structure of metals on the basis of particles is to imagine an array of positively-charged ions sitting in a negatively-charged "gas" of free electrons. Coulombic attraction holds these oppositely-charged particles together, but the positively-charged ions are attracted to negatively charged particles outside the metal as well, such as the negative ions (anions) in an electrolyte. For a given ion at the surface of a metal, there is a certain amount of energy to be gained or lost by dissolving into the electrolyte or becoming a part of the metal, which reflects an atom-scale tug-of-war between the electron gas and dissolved anions. The quantity of energy then strongly depends on a host of variables, including the types of ions in a solution and their concentrations, and the number of electrons present at the metal's surface. In turn, corrosion processes cause electrochemical changes, meaning that they strongly affect all of these variables. The overall interaction between electrons and ions tends to produce a state of local thermodynamic equilibrium that can often be described using basic chemistry and a knowledge of initial conditions.
In a given environment (one standard medium is aerated, room-temperature seawater), one metal will be either more noble or more active than the next, based on how strongly its ions are bound to the surface. Two metals in electrical contact share the same electron gas, so that the tug-of-war at each surface is translated into a competition for free electrons between the two materials. The noble metal will tend to take electrons from the active one, while the electrolyte hosts a flow of ions in the same direction. The resulting mass flow or electrical current can be measured to establish a hierarchy of materials in the medium of interest. This hierarchy is called a Galvanic series, and can be a very useful in predicting and understanding corrosion.
Some metals are more intrinsically resistant to corrosion than others, either due to the fundamental nature of the electrochemical processes involved or due to the details of how reaction products form. For some examples, see galvanic series. If a more susceptible material is used, many techniques can be applied during an item's manufacture and use to protect its materials from damage.
Gold nuggets do not corrode, even on a geological time scale.
The materials most resistant to corrosion are those for which corrosion is thermodynamically unfavorable. Any corrosion products of gold or platinum tend to decompose spontaneously into pure metal, which is why these elements can be found in metallic form on Earth, and is a large part of their intrinsic value. More common "base" metals can only be protected by more temporary means.
Some metals have naturally slow reaction kinetics, even though their corrosion is thermodynamically favorable. These include such metals as zinc, magnesium, and cadmium. While corrosion of these metals is continuous and ongoing, it happens at an acceptably slow rate. An extreme example is graphite, which releases large amounts of energy upon oxidation, but has such slow kinetics that it is effectively immune to electrochemical corrosion under normal conditions.
Given the right conditions, a thin film of corrosion products can form on a metal's surface spontaneously, acting as a barrier to further oxidation. When this layer stops growing at less than a micrometre thick under the conditions that a material will be used in, the phenomenon is known as passivation (rust, for example, usually grows to be much thicker, and so is not considered passivation, because this mixed oxidized layer is not protective). While this effect is in some sense a property of the material, it serves as an indirect kinetic barrier: the reaction is often quite rapid unless and until an impermiable layer forms. Passivation in air and water at moderate pH is seen in such materials as aluminium, stainless steel, titanium, and silicon.
These conditions required for passivation are specific to the material. The effect of pH is recorded using Pourbaix diagrams, but many other factors are influential. Some conditions that inhibit passivation include: high pH for aluminum, low pH or the presence of chloride ions for stainless steel, high temperature for titanium (in which case the oxide dissolves into the metal, rather than the electrolyte) and fluoride ions for silicon. On the other hand, sometimes unusual conditions can bring on passivation in materials that are normally unprotected, as the alkaline environment of concrete does for steel rebar. Exposure to a liquid metal such as mercury or hot solder can often circumvent passivation mechanisms.
Galvanized surface
Plating, painting, and the application of enamel are the most common anti-corrosion treatments. They work by providing a barrier of corrosion-resistant material between the damaging environment and the (often cheaper, tougher, and/or easier-to-process) structural material. Aside from cosmetic and manufacturing issues, there are tradeoffs in mechanical flexibility versus resistance to abrasion and high temperature. Platings usually fail only in small sections, and if the plating is more noble than the substrate (for example, chromium on steel), a galvanic couple will cause any exposed area to corrode much more rapidly than an unplated surface would. For this reason, it is often wise to plate with a more active metal such as zinc or cadmium.
If the environment is controlled (especially in recirculating systems), corrosion inhibitors can often be added to it. These form an electrically insulating and/or chemically impermeable coating on exposed metal surfaces, to suppress electrochemical reactions. Such methods obviously make the system less sensitive to scratches or defects in the coating, since extra inhibitors can be made available wherever metal becomes exposed. Chemicals that inhibit corrosion include some of the salts in hard water (Roman water systems are famous for their mineral deposits), chromates, phosphates, and a wide range of specially-designed chemicals that resemble surfactants (i.e. long-chain organic molecules with ionic end groups).
This figure descender is annodized with a yellow finish. Climbing equipment is available in a wide range of anodized colors.
Aluminium alloys often undergo a surface treatment. Electrochemical conditions in the bath are carefully adjusted so that uniform pores several nanometers wide appear in the metal's oxide film. These pores allow the oxide to grow much thicker than passivating conditions would allow. At the end of the treatment, the pores are allowed to seal, forming a harder-than-usual surface layer. If this coating is scratched, normal passivation processes take over to protect the damaged area.
Cathodic protection (CP) is a technique to control the corrosion of a metal surface by making that surface the cathode of an electrochemical cell.
It is a method used to protect metal structures from corrosion. Cathodic protection systems are most commonly used to protect steel, water, and fuel pipelines and tanks; steel pier piles, ships, and offshore oil platforms.
For effective CP, the potential of the steel surface is polarized (pushed) more negative until the metal surface has a uniform potential. With a uniform potential, the driving force for the corrosion reaction is halted. For galvanic CP systems, the anode material corrodes under the influence of the steel, and eventually it must be replaced. The polarization is caused by the current flow from the anode to the cathode, driven by the difference in electrochemical potential between the anode and the cathode.
For larger structures, galvanic anodes cannot economically deliver enough current to provide complete protection. Impressed Current Cathodic Protection (ICCP) systems use anodes connected to a DC power source (a cathodic protection rectifier). Anodes for ICCP systems are tubular and solid rod shapes of various specialized materials. These include high silicon cast iron, graphite, mixed metal oxide or platinum coated titanium or niobium coated rod and wires.
Passivation is extremely useful in alleviating corrosion damage, but care must be taken not to trust it too thoroughly. Even a high-quality alloy will corrode if its ability to form a passivating film is hindered. Because the resulting modes of corrosion are more exotic and their immediate results are less visible than rust and other bulk corrosion, they often escape notice and cause problems among those who are not familiar with them.
Certain conditions, such as low of oxygen or high of species such as chloride which compete as anions, can interfere with a given alloy's ability to re-form a passivating film. In the worst case, almost all of the surface will remain protected, but tiny local fluctuations will degrade the oxide film in a few critical points. Corrosion at these points will be greatly amplified, and can cause corrosion pits of several types, depending upon conditions. While the corrosion pits only nucleate under fairly extreme circumstances, they can continue to grow even when conditions return to normal, since the interior of a pit is naturally deprived of oxygen. In extreme cases, the sharp tips of extremely long and narrow can cause stress concentration to the point that otherwise tough alloys can shatter, or a thin film pierced by an invisibly small hole can hide a thumb sized pit from view. These problems are especially dangerous because they are difficult to detect before a part or structure fails. Pitting remains among the most common and damaging forms of corrosion in passivated alloys, but it can be prevented by control of the alloy's environment, which often includes ensuring that the material is exposed to oxygen uniformly (i.e., eliminating crevices).
Stainless steel can pose special corrosion challenges, since its passivating behavior relies on the presence of a minor alloying component (Chromium, typically only 18%). Due to the elevated temperatures of welding or during improper heat treatment, chromium carbides can form in the grain boundaries of stainless alloys. This chemical reaction robs the material of chromium in the zone near the grain boundary, making those areas much less resistant to corrosion. This creates a galvanic couple with the well-protected alloy nearby, which leads to weld decay (corrosion of the grain boundaries near welds) in highly corrosive environments. Special alloys, either with low carbon content or with added carbon "getters" such as titanium and niobium (in types 321 and 347, respectively), can prevent this effect, but the latter require special heat treatment after welding to prevent the similar phenomenon of knifeline attack. As its name applies, this is limited to a small zone, often only a few micrometres across, which causes it to proceed more rapidly. This zone is very near the weld, making it even less noticeable1.
Galvanic corrosion occurs when two different metals electrically contact each other and are immersed in an electrolyte. In order for galvanic corrosion to occur, an electrically conductive path and an ionically conductive path are necessary. This effects a galvanic couple where the more active metal corrodes at an accelerated rate and the more noble metal corrodes at a retarded rate. When immersed, neither metal would normally corrode as quickly without the electrically conductive connection (usually via a wire or direct contact). Galvanic corrosion is often utilised in sacrificial anodes. What type of metal(s) to use is readily determined by following the galvanic series. For example, zinc is often used as a sacrificial anode for steel structures, such as pipelines or docked naval ships. Galvanic corrosion is of major interest to the marine industry and also anywhere water can contact pipes or metal structures.
Factors such as relative size of anode (smaller is generally less desirable), types of metal, and operating conditions (temperature, humidity, salinity, &c.) will affect galvanic corrosion. The surface area ratio of the anode and cathode will directly affect the corrosion rates of the materials.
Microbial corrosion, or bacterial corrosion, is a corrosion caused or promoted by microorganisms, usually chemoautotrophs. It can apply to both metals and non-metallic materials, in both the presence and lack of oxygen. Sulfate-reducing bacteria are common in lack of oxygen; they produce hydrogen sulfide, causing sulfide stress cracking. In presence of oxygen, some bacteria directly oxidize iron to iron oxides and hydroxides, other bacteria oxidize sulfur and produce sulfuric acid. Concentration cells can form in the deposits of corrosion products, causing and enhancing galvanic corrosion.
High temperature corrosion is chemical deterioration of a material (typically a metal) under very high temperature conditions. This non-galvanic form of corrosion can occur when a metal is subject to a high temperature atmosphere containing oxygen, sulphur or other compounds capable of oxidising (or assisting the oxidation of) the material concerned. For example, materials used in aerospace, power generation and even in car engines have to resist sustained periods at high temperature in which they may be exposed to an atmosphere containing potentially highly corrosive products of combustion.
The products of high temperature corrosion can potentially be turned to the advantage of the engineer. The formation of oxides on stainless steels, for example, can provide a protective layer preventing further atmospheric attack, allowing for a material to be used for sustained periods at both room and high temperature in hostile conditions. Such high temperature corrosion products in the form of compacted oxide layer glazes have also been shown to prevent or reduce wear during high temperature sliding contact of metallic (or metallic and ceramic) surfaces.
10. Stress Corrosion Cracking
Stress Corrosion Cracking (SCC) requires the coexistence at a surface of a tensile stress and an aggressive environment. Stress corrosion cracks are seldom discrete but usually take the form of a network of branching cracks which propagate round or across grains in a direction normal to stress which may be that applied by design (magnified by stress concentration), may arise from differential thermal expansion, from fabrication, (mechanical working, welding or a combination of heat treatment and machining) or from strain caused by the accumulation of corrosion products. There is usually an incubation period which may be of long duration between the time at which the conditions required for stress corrosion are realised and the start of cracking.
Treshold stress Treshold stress
There is, in many cases, a threshold stress (or more properly a threshold stress intensity which is a function of stress and defect size and sharpness) below which cracking will not occur within the economic life of a design or the economic time for carrying out an experiment.
Initiation and Times to failure
The relationships outlined above govern probabilities. One specimen may survive arduous conditions for a longer period than another in relatively easy conditions. A component may last for months or even years without apparent deterioration, but fail in a few days after cracking starts. On the other hand a crack may progress for a substantial distance (in the order of centimetres) and then slow down or stop, presumably because the corroding medium can no longer gain access to the tip of the crack or because the crack has relieved the stress which initiated it. The relationships outlined above govern probabilities. One specimen may survive arduous conditions for a longer period than another in relatively easy conditions. A component may last for months or even years without apparent deterioration, but fail in a few days after cracking starts. On the other hand a crack may progress for a substantial distance (in the order of centimetres) and then slow down or stop, presumably because the corroding medium can no longer gain access to the tip of the crack or because the crack has relieved the stress which initiated it.
Susceptible materials / Specific environments
It is probable that every solid material is subject to stress corrosion by some environment, liquid, gas, or even solid which may be specific to it. Plastics suffer stress corrosion by organic liquids and ceramics by atmospheric moisture. "Season Cracking" of brass is stress corrosion by ammonia and 'Liquid Metal Penetration" stress corrosion by a liquid metal.
The most common occurrences in process plant are "Caustic Embrittlement", the stress corrosion of steels by caustic soda solutions which concentrate in cavities or crevices, and stress corrosion of steels and nickel alloys by halide, usually chloride, solutions and by polythionic acids. It is probable that every solid material is subject to stress corrosion by some environment, liquid, gas, or even solid which may be specific to it. Plastics suffer stress corrosion by organic liquids and ceramics by atmospheric moisture. "Season Cracking" of brass is stress corrosion by ammonia and 'Liquid Metal Penetration" stress corrosion by a liquid metal.
Stress corrosion of austenitic steels is usually transgranular. When this is the case the attack has a very characteristic branching habit which is easily recognisable in SEM fractographic and metallographic section examination.
Some forms of stress corrosion cracking (including some instances of chloride attack on austenitic steels) are intergranular and aggravated by "Sensitisation".
It may not be possible to distinguish between this type of stress
corrosion, intergranular penetration and hydrogen penetration which arises from corrosion by an aqueous electrolyte. None of these mechanisms will proceed in the absence of liquid water. Even if the presence of liquid water is not at first sight evident, a rigorous examination of the conditions will usually reveal a condensation or droplet transfer process which deposits a liquid aqueous solution of alkali or alkaline earth halides and hydroxides and concentrates it by evaporation.
The most common forms of stress corrosion cracking can be prevented by eliminating liquid water and where possible by excluding aggressive agents such as hydroxides, chlorides and thionic acids. Stress should be reduced, and sensitisation avoided. The tendency to stress corrosion cracking of austenitic steels can be reduced by increasing the nickel content. The addition of molybdenum may retard initiation.
Hydrogen in damaging concentrations in metal can lead, in smaller quantities to "Hydrogen Penetration" and in larger quantities to "Hydrogen Degradation". Pickling without the correct inhibitor, plating under conditions which cause hydrogen to be evolved, welding in wet conditions and/or with damp welding rods other than the low hydrogen type and exposure to an environment such as H2S that generates atomic hydrogen at the surface can also introduce hydrogen. Of course, corrosion can generate environments conducive to hydrogen embrittlement.
12. Corrosion Fatigue
Corrosion may prevent, retard, accelerate and/or initiate fatigue cracking. For a description of the latter phenomenon: see Fatigue
Corrosion phenomena may either be involved in the initiation, or in the propagation of the fatigue cracking.
Crack Blunting
Crack blunting occurs when the potential conditions in the environment at the root of a fatigue crack are such as to attack the material at right angles to the crack, and thereby stop, or reduce the rate of, penetration. The result of eliminating from an environment a supposedly aggressive constituent which had previously suppressed fatigue crack propagation can have disconcerting results. For example, substituting an inert atmosphere such as vacuum or an inert gas for a carbon dioxide environment very significantly increases the incidence of fatigue cracking in magnesium.
Environments – Overview
Environmental cracking is a pattern of behavior, a set of symptoms, rather than a single mechanism. Such cracking process occurs in pure metals and in all alloy systems, from aluminum to zirconium. Environments as mild as moist air and as harsh as mixed acids can cause cracking.
In general, environmental cracking occurs when a critical combination of temperature, tensile stress, metallurgical structure and environmental conditions coincide. For any given alloy-environment combination, the engineering parameters of concern are as follows:
1. Threshold stresses above which cracking occurs;
2. Metallurgical variables (heat treatment, structure, cold work) which render the alloy susceptible;
3. Environmental boundary conditions for cracking such as temperature, solution composition, pH, electrode potential, necessary impurities, etc...
In this information system the approach will largely be limited to empirical observations of the boundary conditions within which cracking occurs. From an engineering standpoint, an understanding of these conditions can reduce the need for a satisfactory theoretical explanation (which often does not fully exist) in that environmental cracking can be controlled by controlling these conditions. Environmental cracking is a pattern of behavior, a set of symptoms, rather than a single mechanism. Such cracking process occurs in pure metals and in all alloy systems, from aluminum to zirconium. Environments as mild as moist air and as harsh as mixed acids can cause cracking.
In general, environmental cracking occurs when a critical combination of temperature, tensile stress, metallurgical structure and environmental conditions coincide. For any given alloy-environment combination, the engineering parameters of concern are as follows:
1. Threshold stresses above which cracking occurs;
2. Metallurgical variables (heat treatment, structure, cold work) which render the alloy susceptible;
3. Environmental boundary conditions for cracking such as temperature, solution composition, pH, electrode potential, necessary impurities, etc...
In this information system the approach will largely be limited to empirical observations of the boundary conditions within which cracking occurs. From an engineering standpoint, an understanding of these conditions can reduce the need for a satisfactory theoretical explanation (which often does not fully exist) in that environmental cracking can be controlled by controlling these conditions.
Aqueous Chlorides
Caustic Environments
Ammoniacal Environments
Nitrogenous Environments
Sulfurous Environments
High-Purity Water and Steam
Source: http://www.bu.edu.eg/portal/uploads/Engineering,%20Shoubra/Civil%20Engineering/2469/crs-6270/Files/corrosion.doc
Web site to visit: http://www.bu.edu.eg
Author of the text: indicated on the source document of the above text
If you are the author of the text above and you not agree to share your knowledge for teaching, research, scholarship (for fair use as indicated in the United States copyrigh low) please send us an e-mail and we will remove your text quickly. Fair use is a limitation and exception to the exclusive right granted by copyright law to the author of a creative work. In United States copyright law, fair use is a doctrine that permits limited use of copyrighted material without acquiring permission from the rights holders. Examples of fair use include commentary, search engines, criticism, news reporting, research, teaching, library archiving and scholarship. It provides for the legal, unlicensed citation or incorporation of copyrighted material in another author's work under a four-factor balancing test. (source: http://en.wikipedia.org/wiki/Fair_use)
The information of medicine and health contained in the site are of a general nature and purpose which is purely informative and for this reason may not replace in any case, the council of a doctor or a qualified entity legally to the profession.
The texts are the property of their respective authors and we thank them for giving us the opportunity to share for free to students, teachers and users of the Web their texts will used only for illustrative educational and scientific purposes only.
All the information in our site are given for nonprofit educational purposes